Lange Zeit glaubten Ärzte und Forscher das die erste in der medizinischen Literatur beschriebene Person mit dem was wir heute “Primordialer Kleinwuchs” nennen CAROLINE CRACHAMI war: bekannt auch als “Sicilian Dwarf” – geboren 1815 in Sizilien.
Der Besuch einer unserer betroffenen deutschen Familien im Herzog Anton Ulrich-Museum in Braunschweig im Jahr 2019 brachte jedoch eine verblüffende Tatsache ans Licht – die tatsächlich bis dahin noch NIEMAND in den richtigen Kontext gebracht hatte. Das Museum zeigt die Wachsfigur von
NICOLAS FERRY
-Geboren am 14.10.1741 (!!) in Frankreich
-Geburtsgewicht: 625 Gramm
-Körperlänge: 21 Zentimeter
-Gestorben am 08.06.1764 an den Folgen einer Erkältung
Aufgrund unseres Hinweises hat der führende Forscher (Dr. Michael Bober, USA) alle Fakten und Bilder begutachtet und ist sicher das Nicolas Ferry an einer Form von Primordialem Kleinwuchs litt – an welcher ist momentan allerdings noch nicht abschließend geklärt.
Fakt ist aber das es umfangreiche Aufzeichnungen über das Leben des “Bebe” genannten Franzosen gibt: so wissen wir zum Beispiel das er mit 18 Monaten zu sprechen begann und mit 2 Jahren im Stande war zu laufen. Auch wurde nach seinem Tod eine umfassende Obduktion vorgenommen bei welcher speziell die Knochen- und Hirnanomalien sehr detailliert beschrieben wurden.
Fakt ist auch das die Forschung nun mit Sicherheit neue Erkenntnisse zum Krankheitsbild von Nicolas Ferry gewinnen kann – und das alles nur, weil die “richtige” Familie mit offenen Augen durchs Museum gegangen ist!
 Unser Dank geht ans “Herzog Anton Ulrich-Museum Braunschweig” für die Erlaubnis zur Veröffentlichung des Fotos und auch für die Unterstützung bei Recherchen zum Leben von Nicolas Ferry sowie die zur Verfügung gestellten Unterlagen seiner Autopsie.
Unser Dank geht ans “Herzog Anton Ulrich-Museum Braunschweig” für die Erlaubnis zur Veröffentlichung des Fotos und auch für die Unterstützung bei Recherchen zum Leben von Nicolas Ferry sowie die zur Verfügung gestellten Unterlagen seiner Autopsie.

CAROLINE CRACHAMI
CAROLINE CRACHAMI – besser bekannt als “Sicilian Dwarf”.
-Geboren 1815 in Sizilien
-Geburtsgewicht: 450 Gramm
-Körperlänge: 20 Zentimeter
Ab dem Jahr 1824 wurde sie in einem Zirkus als “Kuriosität” ausgestellt.
Gestorben ist sie im Jahr 1824 an den Folgen einer Tuberkulose. Zum Zeitpunkt ihres Todes war sie 57 cm groß und wog zwischen 2-3 Kilogramm.
Ihre Knochen sind heute im “Hunterian Museum at the Royal Collage of Surgeons” in Glasgow ausgestellt.
ANDERE HISTORISCHE PERSÖNLICHKEITEN MIT PRIMORDIALEM KLEINWUCHS

General Mite
Im Alter von 16 Jahren war er 53cm groß und 4 Kilogramm schwer.

Prince Nicholi
Im Alter von 32 Jahren war er 66cm groß und 7 Kilogramm schwer.
ENTDECKUNG UND FORSCHUNG
WAS IST PRIMORDIALER KLEINWUCHS?
Dem Wortstamm nach bedeutet
PRIMORDIAL: Merkmal des frühesten Entwicklungsstadiums eines Organismus – “ursprünglich” und vor der Geburt.
und
KLEINWUCHS: unproportional und von kleiner Statur.
DR HELMUT SECKEL
-Geboren im Jahr 1900 in Deutschland
-Studierte Medizin an der Universität von Berlin und spezialisierte sich auf Kinderheilkunde
-1936 floh er vor dem Nasziregime in die USA
-Er wurde Professor der Kinderheilkunde an der “University of Chicago Medical School”
-Vor seinem Tod im Jahr 1960 veröffentlichte er im gleichen Jahr eine Abhandlung mit dem Titel: “Vogelköpfige Zwerge: Entwicklungslehre inklusive Betrachtung der menschlichen Proportionen”. Nach ihm und seinen Erkenntnissen benannt sprechen wir noch heute vom
SECKEL-SYNDROM
Stellt sich dar durch
-“schwere intrauterine Wachstumsretadierung”, oder auf deutsch: vermindertes Wachstum schon in der Schwangerschaft
-auch nach der Geburt vermindertes Wachstum
-schwere unverhältnismässige Mikrozephalie (extrem kleiner Kopf) von Geburt an
-“Vogelartiges” Gesichtsprofil
-andere Anomalien wie zum Beispiel:
fliehende Stirn, große und flache Nase, fliehender Unterkiefer und Kinn

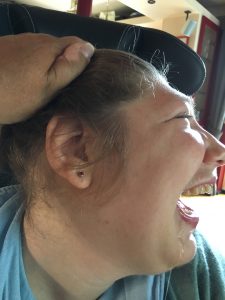
(Franziska Anton, Deutschland)
DR. FRANK MAJEWSKI
-geboren 1941 in Deutschland
-studierte Medizin in Saarbrücken, Freiburg, Wien und Münster und promovierte 1967 am Institut für Humangenetik in Münster
-Spezialisierung auf Kinderheilkunde am Universitätsklinikum für Kinder- und Jugendmedizin in Tübingen
-1978 wurde er als Professor für Humangenetik und Kinderheilkunde an die Universität nach Düsseldorf berufen, wo er bis zu seinem Tod im Jahr 2001 arbeitete.
-im Mai 1982 veröffentlichte er im “American Journal of Medical Genetics” einen Artikel mit dem Titel “Studie über Mikrozephalen Primordialen Kleinwuchs II: über die Knochenfehlbildungen bei Typ II des Primordialen Kleinwuchses”. Diese Veröffentlichung war der Beginn von allem was wir heute wissen über
MOPD Typ 1 +3/ MOPD Typ 2
MOPD Typ 1+3: Bereits 1967 beschrieben die beiden Kinderradiologen Hooshang Taybi und David Linder eine Form von angeborenem Kleinwuchs mit Fehlbildungen der Knochen. Im Jahr 1976 und (mit seiner Veröffentlichung im AJMG) 1982 definierte Dr Majewski diese Form des Kleinwuchses als MOPD Typ 1/Typ 3.
Auffallend dabei war und ist: Mikrozephalie (extrem kleiner Kopf) steht IMMER IN ZUSAMMENHANG mit Osteodysplasien (Knochenfehlbildungen).
Diese Knochenfehlbildungen können zum Beispiel auftreten an
-den Epiphysen (den Knochenenden): diese sind oftmals nicht “rund”, sondern eher kantig/eckig/mit Ausbuchtungen
-den Metaphysen (dem Knochenschaft): dieser hat dann kein “glattes” Ende, sondern eher ein ausgefranstes/eckiges/kantiges
-den Spinalkanälen (Rückenmarkskanal): dieser ist dann im Querschnitt nicht rechteckig, sondern unförmig/irregulär
Diese Merkmale treten bei einem betroffenen Patienten nicht zwingend alle gleichzeitig auf. Was aber allen Betroffenen gemein ist, sind die stark verkürzten Oberarm-, Unterarm-, Oberschenkel- und Unterschenkelknochen.
Diese Form des Kleinwuchses wird ausschließlich vererbt. Und zwar “autosomal-rezessiv” (nur wenn beide Eltern es in sich tragen).
Unterteilte man zu Beginn die Patienten noch in die Formen MOPD Typ 1 UND MOPD Typ 3 stellte man Ende der 80er Jahre fest das diese beiden Formen so geringe Abweichungen voneinander haben das man sie “zusammenfassen” kann: heute gibt es nur noch MOPD Typ 1.

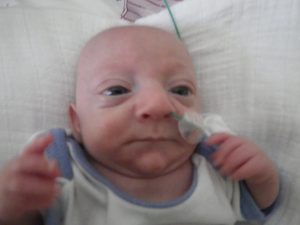
(Links: Lotta; rechts: Jonathan – beide aus Deutschland)
MOPD Typ 2: wurde durch die im Jahr 1982 veröffentlichte Studie bekannt.
Im Gegensatz zu Typ 1 sind von Typ 2 Betroffene geistig oftmals vollkommen normal entwickelt, sie haben mit (heutzutage) über 30 Jahren eine längere Lebenserwartung, bleiben aber körperlich sehr viel kleiner. Laut einer Studie der Universität Erlangen aus dem Jahr 2008 werden Betroffene nicht größer als 1 Meter.
Genau wie bei Typ 1 haben diese Menschen Knochenfehlbildungen und einen extrem kleinen Kopf.
Im Laufe der Zeit hat sich herausgestellt das Patienten mit MOPD Typ 2 zu Aneurysmen (eine Erweiterung der Gefäßwand, hier vorwiegend im Gehirn) neigen. Forschungsergebnisse zeigen, das 100% aller Patienten die das Alter von 24 Jahren erreichen, ein Aneurysma haben/hatten. Ebenfalls besteht bei Betroffenen die Neigung zu Moyamoya: einer Erkrankung bei der es zur Verengung oder zum Verschluß von Hirnaterien kommt, was dann zum Hirninfarkt oder zu Hirnblutungen führen kann.
Seit man dieses Wissen erlangt hat: werden bei Patienten mit MOPD Typ 2 regelmäßige MRT-Aufnahmen des Gehirns durchgeführt um möglichst schnell auf Probleme reagieren zu können.
Als medizinische Probleme konnten ebenfalls Niereninsuffizienz und Bluthochdruck identifiziert werden.

(Lina aus Deutschland)
ROIFMAN-SYNDROM
-erstmals beschrieben im Jahr 1997
-charakteristische Merkmale sind unter anderem das Fehlen von Antikörpern im Blut, geistige Behinderung, Gesichtsfehlbildungen mit Mikrozephalus, verzögertes Wachstum (vor und nach der Geburt) und Muskelhypotonie (Muskelschwäche).
MOPD Typ 1, Roifman-Syndrom und das “Lowry-Wood-Syndrome” (über das mir zum aktuellen Zeitpunkt keine weiteren Informationen vorliegen) werden durch “Fehler” auf demselben Gen hervorgerufen. Deswegen sollte an dieser Stelle gesagt sein das sich manche Patienten in “Grauzonen” bewegen: sie haben Symptome die auf mehrere dieser Gendefekte zutreffen – was eine genaue Diagnosestellung sehr viel schwieriger macht.
Durch das verursachende Gen ist aber, egal welcher dieser drei Gendefekte es schlußendlich ist, das Immunsystem gestört. Das äußert sich zum Beispiel dadurch das:
-die Impf-Antikörper in der Regel sehr niedrig sind
-die meisten Patienten nicht in der Lage sind spezielle Antikörper zu produzieren
-zirkulierende B-Zellen (weiße Blutkörperchen) eher am unteren Ende der meßbaren Skala angesiedelt sind, wohingegen abgelagerte B-Zellen im Normalbereich liegen
-Anzahl und Funktion von T-Zellen im normalen Bereich liegen, aber CD8 +T-Zellen stark minimiert sind bei
MOPD Typ 1.
Außerdem sind Hirnfehlbildungen bekannt:
-glattes Gehirn ohne Hirnwindungen
-teilweises oder komplettes Fehlen des Balken, der die linke und rechte Hirnhälfte verbindet
-unterentwickelter Frontallappen
EMPFEHLUNGEN
Aus allen diesen Erkenntnissen konnten einige Empfehlungen für Patienten mit einer dieser Formen von Primordialem Kleinwuchs gewonnen werden:
-regelmäßiges MRT des Gehirns
-Echokardiogram und Ultraschall des Bauches
-regelmäßige Kontrolle des Immunsystems
-Augenärztliche Kontrolle
-Orthopädische Kontrolle
-Aufmerksamkeit und Kontrolle bei Fütterung und Gewichtszunahme
ZEITSTRAHL
1960: erste Beschreibung des Seckel-Syndroms durch Dr Helmut Seckel
1967: die Kinderradiologen Hooshang Taybi und David Linder beschreiben zum ersten Mal eine Form von Kleinwuchs die sich später als MOPD Typ 1 herausstellen sollte
1976: Beschreibung von MOPD Typ 1 durch Dr Frank Majewski und seine Kollegen
1982: Veröffentlichung einer Studie über MOPD Typ 2 durch Dr Frank Majewski und seine Kollegen
1997: erste Beschreibung des Roifman-Syndroms
2000: erstes Treffen Betroffener in den USA
2008: Zusammenarbeit der Forscher Dr Bober (USA), Dr Jackson (Schottland) und ihrer Teams startet
2008: die WALKING WITH GIANTS FOUNDATION in Liverpool wird gegründet, sie steht von Anfang an in engem Kontakt mit dem Forschungsteam
2008-heute: das Forschungsteam konnte durch seine Arbeit mittlerweile 10 verschiedene Formen/Unterformen von Primordialem Kleinwuchs identifizieren.
Zu dieser Identifikation gehört mehr als nur “dem Kind einen Namen zu geben”!
LIGASE 4: Erst seit wenigen Jahren ist die Form LIGASE 4 bekannt (nach aktuellen Schätzungen leben aktuell nur 30 Menschen WELTWEIT damit). Zu Beginn wusste man noch nicht das diese Kinder im Laufe ihres Lebens IMMER eine Stammzellenspende benötigen, einige sind verstorben da nicht rechtzeitig geholfen werden konnte. Dank der Forschung hat man dieses Wissen heute jedoch und kann schon rechtzeig beginnen einen Stammzellenspender zu suchen und so das Leben dieser Kinder retten.
MOPD Typ2: Ebenfalls erst in den letzten Jahren hat die Forschung herausgefunden das Menschen mit MOPD Typ2 IMMER vor Erreichen des 24.Lebensjahres an Aneurysmen erkranken, manche sogar mehrfach. Diese Erkenntnis führte dazu das Betroffene heute alle 6 Monate ein MRT des Kopfes erhalten. So können Aneurysmen frühzeitig erkannt und behandelt werden. Sind noch vor 15 Jahren Betroffene gestorben weil unerkannte Aneurysmen rupturierten, sind heute die Überlebenschancen durch die frühzeitigen Kontrollen sehr viel höher.
MOPD Typ1: Seit weniger als 10 Jahren ist dank der Forschung bekannt das man das Immunsystem von Betroffenen durch die regelmäßige Gabe von “Immunglobulinen” unterstützen kann. Die Infektanfälligkeit wird reduziert und dadurch die Lebenserwartung erhöht.
WIE VIELE BETROFFENE GIBT ES ??
Auf diese Frage gibt es KEINE 100% richtige Antwort: einige Betroffene lassen sich nicht in der Datenbank der Forscher registrieren – einige Betroffene haben vielleicht noch gar keine Diagnose – einige Betroffene leiden vielleicht auch an einer Form die noch nicht beschrieben wurde. Es gibt also nur Schätzungen.
Diese belaufen sich auf circa 500 Menschen weltweit.
Fakt ist, das im Jahr 2018 in der Datenbank 114 Betroffene (63 Frauen und 51 Männer) registriert waren – WELTWEIT.
20% der Betroffenen haben noch KEINE genaue Diagnose erhalten: die Ärzte sprechen hier von “PD unknown” (also unbekannte Form). Diese Zahl macht sehr deutlich das die Forschung noch einen langen Weg und viel Arbeit vor sich hat! Eine Diagnose zu erhalten kann für Betroffene Lebensnotwendig sein – wie bei der Diagnose LIGASE 4.
QUELLEN:
-Präsentation von Michael B. Bober, M.D, Ph.D vom 15.08.2018
-WIKIPEDIA
-Fotos mit Urheberrecht (von Stefanie Mihm, Barbara Anton, Edina V., Jasmin W. und Simone Braunsdorf-Kremer)
Mai 2021:
Das Forschungsteam um Dr. Michael Bober hat eine Publikation über MOPD Typ2 veröffentlicht
https://ojrd.biomedcentral.com/articles/10.1186/s13023-021-01852-y
Dezember 2021:
Das Forschungsteam um Dr. Michael Bober hat eine weitere Publikation über MOPD Typ2 veröffentlicht
https://www.ncbi.nlm.nih.gov/books/NBK575926/pdf/Bookshelf_NBK575926.pdf